
Tic-tac, 1 mois avant la première date butoir de la transition règlementaire
Legacy devices, où en êtes-vous dans le calendrier ?
Date de publication
25/04/2024
Legacy devices, où en êtes-vous dans le calendrier ?
Date de publication
25/04/2024
Contexte actuel
Avant l'entrée en vigueur des nouveaux règlements européens relatifs aux dispositifs médicaux (MDR 2017/745 et IVDR 2017/746), la législation applicable était régie par deux directives : la directive 93/42/CEE pour les dispositifs médicaux et la directive 90/385/CEE pour les dispositifs médicaux implantables actifs.
Ces directives définissaient les normes harmonisées et la législation à respecter pour mettre votre dispositif médical sur le marché en Europe. Les fabricants devaient soumettre leurs dispositifs à une évaluation de la conformité par un organisme notifié, qui était chargé de vérifier que les dispositifs répondaient aux exigences des directives.
Les nouveaux règlements ont pour objectif de proposer une classification, et d’améliorer la sécurité, la performance, la traçabilité, la surveillance après-commercialisation, ainsi que d’harmoniser ces exigences pour les dispositifs médicaux au sein de l’Union européenne.
Cette nouvelle réglementation est assortie d'une période de transition bien définie, visant à préserver la continuité des soins pour les patients et à éviter tout risque de pénurie des dispositifs médicaux.
La période de transition
La transition vers la nouvelle réglementation dans le domaine des DM représente un défi majeur pour l’écosystème ainsi que pour les organismes notifiés chargés de la certification. La nécessité d’assurer la conformité des dispositifs tout en maintenant la continuité de leur mise sur le marché a conduit les autorités européennes à revoir et à ajuster la période de transition en s’adaptant aux évolutions de l’écosystème.
Suspendre temporairement l'utilisation des dispositifs médicaux certifiés conformément aux anciennes directives, en attendant leur mise en conformité avec cette nouvelle règlementation aurait engendré des risques de pénurie et une atteinte à la santé publique.
En ce sens, la Commission européenne a prévu une période de transition afin de permettre aux fabricants de mettre en conformité leurs dispositifs aux nouvelles exigences réglementaires sans impacter leur mise sur le marché.
Frise chronologique : les phases de la transition règlementaire
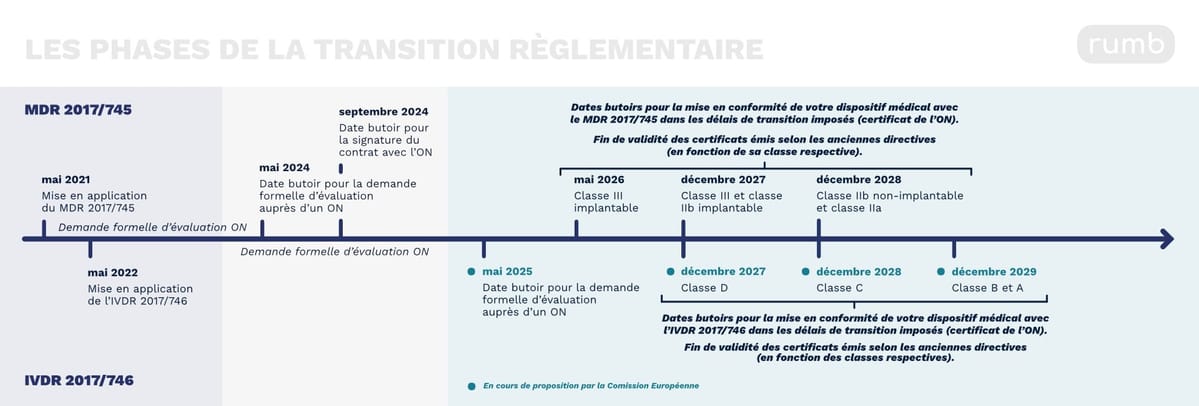
Pour les DM (MDR 2017/745) :
Pour les DMDIV (IVDR 2017/746), le calendrier de la transition est encore en cours de validation auprès des instituions européennes. Voici les grandes lignent qui se dégage dans la proposition de la Commission Européenne (février 2024) :
Période de transition mais sous quelles conditions ?
L’application de cette extension de la période transitoire est conditionnelle.
Fabricants de Legacy Devices vous devez remplir les conditions suivantes pour bénéficier de cette période de transition :
• Votre DM doit être maintenu conforme aux anciennes directives
• Les produits ne doivent pas subir de modifications substantielles (conception ou utilisation prévue)
• Les DM ne présentent pas de risque inacceptable pour la santé ou les utilisateurs
Les fabricants doivent avoir effectuer une demande d’évaluation formelle auprès d’un organisme notifié et avoir mis en place un système de gestion de la qualité conforme aux exigences règlementaires (comprenant la vigilance, la surveillance après commercialisation et l’enregistrement des acteurs économiques sur EUDAMED) : MDR pour les DM (au plus tard le 26 mai 2024) et l’IVDR pour les DMDIV (possiblement avant le 26 mai 2025).
de transition
Le défi clinique
L’introduction d’une nécessité de validation clinique pour les DM est une barrière à l’entrée conséquente, notamment pour les DM legacy encore sous directive.
Le changement de paradigme introduit par les règlements européens quant au renforcement des exigences cliniques va amener les entreprises à devoir repenser leur stratégie clinique et fort probablement à conduire des investigations cliniques pour démontrer, par des preuvres cliniques suffisantes, le rapport bénéfice/risque de leur produit.
La validation des preuves cliniques est un pré-requis pour obtenir le droit de continuer à commercialiser un produit en Europe (marquage CE). Au regard du calendrier de la transition règlementaire, il est encore temps d'anticiper ces questions pour les fabricants.
Quelques statistiques

Si vous vous reconnaissez dans ces chiffres, c’est peut-être que vous avez manqué de ressources et d’expertise ciblée à un moment stratégique de la vie de votre entreprise.
Vous avez formulé votre demande d’évaluation de la conformité auprès d’un ON mais vous manquez de visibilité ou de sérénité sur les prochaines échéances de commercialisation de votre DM ?
Notre équipe pourrait vous éclairer.
Cet article est diffusé à titre informatif et ne constitue pas une référence normative ou règlementaire.